
 1005 Alcyon Dr Bellmawr NJ 08031
1005 Alcyon Dr Bellmawr NJ 08031

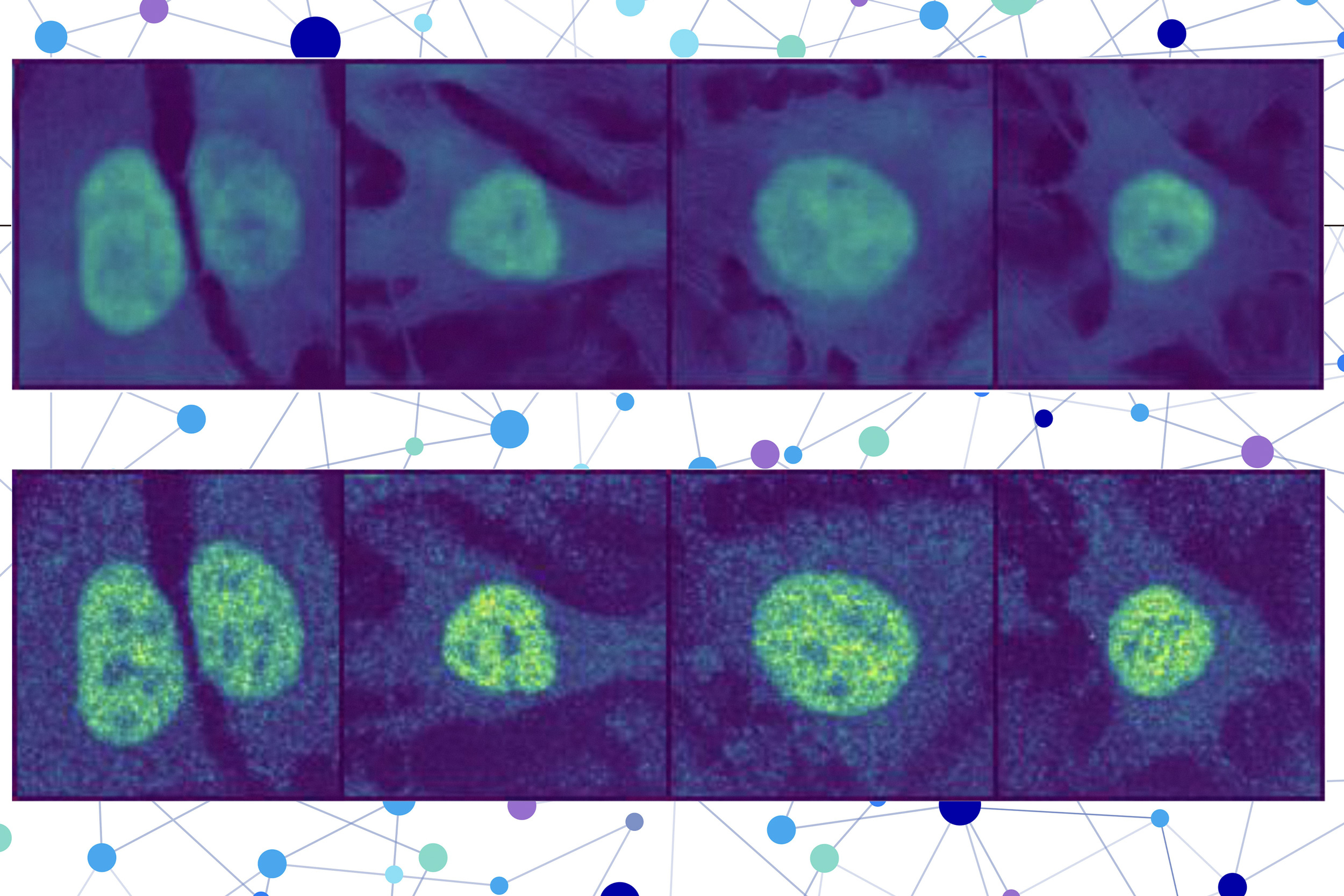
Proteins located in the wrong parts of the cell can cause a variety of diseases such as Alzheimer’s disease, cystic fibrosis, and cancer. However, there are about 70,000 different proteins and protein variants in a single human cell, and since scientists usually only test a few in one experiment, manually identifying the location of proteins is very expensive and time-consuming.
A new generation of computing technologies attempt to simplify the process using machine learning models that typically utilize datasets containing thousands of proteins and their locations, measured by multiple cell lines. One of such data sets is the Human Protein Atlas, which categorizes the subcellular behavior of more than 13,000 proteins in more than 40 cell lines. However, despite this, the Human Protein Atlas explored only 0.25% of all possible pairings for all proteins and cell lines in the database.
Now researchers at MIT, Harvard University and the extensive institutes at MIT and Harvard have developed a new computational method that can effectively explore the rest of unknown spaces. Their method can predict the location of any protein in any human cell line, even if the protein and cells have never been tested.
By localizing proteins at the single-cell level, rather than as an average estimate in all cells of a particular type, their technique goes a step further than many AI-based approaches. For example, such single-cell localization can identify the location of proteins in specific cancer cells after treatment.
The researchers combined protein language models with a special type of computer vision model to capture rich details about proteins and cells. Finally, the user receives an image of a cell with a highlighted section indicating the model’s prediction of where the protein is located. Because protein localization indicates its functional status, the technology can help researchers and clinicians more effectively diagnose disease or identify drug targets, while also allowing biologists to better understand how complex biological processes relate to protein localization.
“You can do these protein localization experiments on your computer without touching any lab’s desk, hoping to save months of effort. While you still need to validate the predictions, this technique may be like the initial screening of experimental tests.”
TSEO is added to paper from the Graduate Students of the Department of Electrical Engineering and Computer Science (EEC) and the Eric and Wendy Schmidt Centers of the Broad Institute. Yunhao Bai of Kuankai Institute; and Assistant Professor of the Advanced Institute, a member of the Broad Institute and Professor of Engineering of Caroline Uhler, Andrew and Erna Viterbi and Senior Author of MIT Data, Systems and Society (IDSS), Fei Chen, also head of ERIC and Wendys Schmidt Center and Wendys Schmidt Center, as well as Director of MIT Data, Systems and Society (IDSS) and Dikistss (MIT and Wendy Schmidt Center) of MIT and Wendy Schmidt Center. The study appears today Natural Method.
Collaborative Model
Many existing protein prediction models can only predict based on trained protein and cellular data, or cannot pinpoint the location of proteins in a single cell.
To overcome these limitations, the researchers created a two-part method to predict subcellular locations of unseen proteins, called pups.
The first part uses protein sequence models to capture the localization of proteins and their 3D structures based on the amino acid chains forming IT.
The second part combines an image introduction model, which is designed to fill in missing parts of the image. The computer vision model looks at stained images of three cells to collect information about the cell, such as its type, individual characteristics, and whether it is under stress.
PUPS uses an image decoder output to show the highlighted image of the predicted position to predict where each model creates a representation of the protein to predict where the protein is in a single unit.
“Different cells in cell lines exhibit different characteristics, and our model is able to understand this nuance,” Tseo said.
User input sequences of amino acids that form proteins and three cell stained images – one for nucleus, one for microtubules, and one for endoplasmic reticulum. Then the cubs remain.
A deeper understanding
The researchers used some tips during the training to teach the pup how to combine information in the way each model so that it can make educated guesses about the protein’s location, even if it has never seen the protein before.
For example, they assigned a secondary task to the model during training: to explicitly name the localization chamber, such as the nucleus. This is done with the main introductory task to help the model learn more effectively.
A good metaphor might be a teacher asking their students to draw all the parts of the flower besides writing their names. This additional step was found to help the model improve its general understanding of possible cellular compartments.
Furthermore, the fact that young trained proteins and cell lines trained can help it gain a deeper understanding of the locations in cellular image proteins that tend to be localized.
The pups can even understand by themselves how different parts of the protein sequence contribute their overall positioning separately.
“Most other methods usually require you to stain the protein first, so you’ve seen it in the training data. Our approach is unique because it can span both protein and cell lines at the same time,” Zhang said.
Since pups can be generalized as invisible proteins, it can capture localization changes driven by unique protein mutations not included in the human protein map.
The researchers confirmed that pups can predict the subcellular location of novel proteins in cell lines by performing laboratory experiments and comparing results. Additionally, pups showed less prediction error on average in the tested proteins compared to the baseline AI method.
In the future, researchers hope to enhance puppies so that models can understand protein-protein interactions and make localization predictions for multiple proteins in cells. In the long run, they want to make predictions in the living human tissues rather than cultured cells.
The study was conducted by the Eric and Wendy Schmidt Center of the Broad Institute, the National Institutes of Health, the National Science Foundation, the Burroughs Welcome Fund, the Searle Scholars Fund, the Searle Scholars Foundation, the Harvard Stem Cell Institute, the Melkin Institute, the Naval Institute, the Naval Institute, and the Department of Energy.