
 1005 Alcyon Dr Bellmawr NJ 08031
1005 Alcyon Dr Bellmawr NJ 08031

In the past – the past day – the task of designing materials is laborious. Over more than 1,000 years, investigators have tried to make gold by combining things like lead, Mercury and sulfur, which they hope they want to be the right proportions. Even famous scientists like Tycho Brahe, Robert Boyle, and Isaac Newton have tried the fruitless efforts we call “alchemy.”
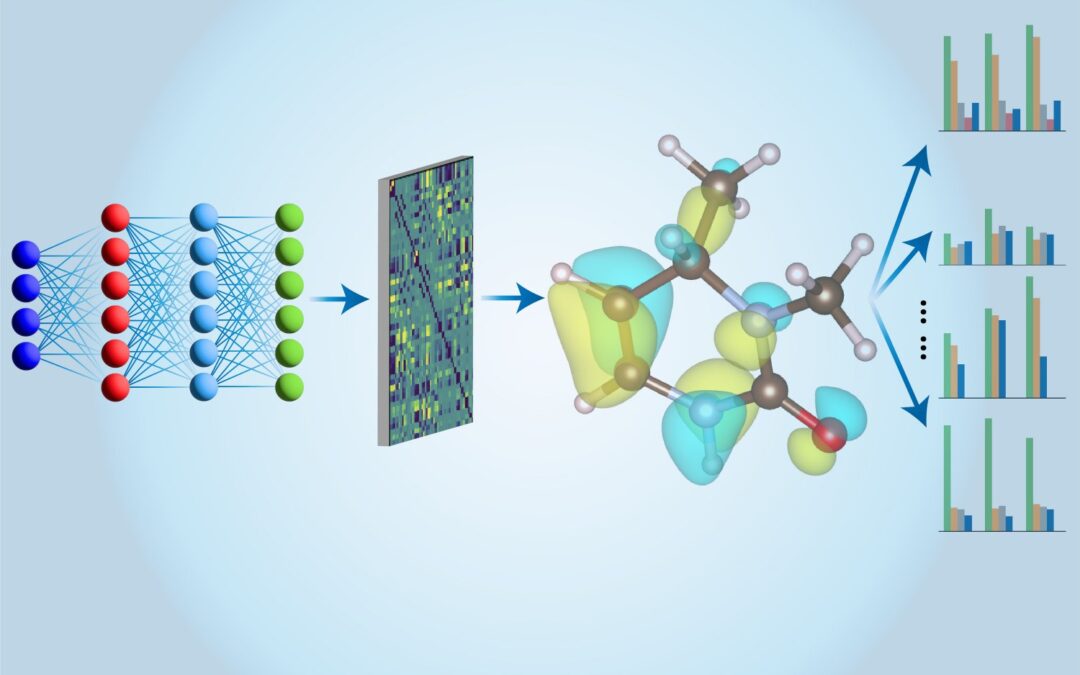
Materials science has certainly come a long way. Over the past 150 years, researchers have benefited from the list of elements to be borrowed, which tells them that different elements have different properties and that one cannot magically transform into another. Furthermore, over the past decade or so, machine learning tools have greatly improved our ability to determine the structural and physical properties of various molecules and matters. New research by the group led by Ju Li, a professor of nuclear engineering at Tokyo Electric Power and professor of materials science and engineering at MIT, offers hope that a significant leap in the ability to design materials is expected to facilitate. Their findings are released in December 2024 Natural Computational Science.
Currently, most machine learning models used to characterize molecular systems are based on density function theory (DFT), which provides a quantum mechanical method that determines the total energy of a molecule or crystal by looking at the electron density distribution, which is basically the average of the average electron volume at each given point located near the molecule. (Walter Kohn co-invented the theory 60 years ago, and he won the Nobel Prize in Chemistry in 1998.) While the method is very successful, it has some shortcomings, according to Lee: “First of all, the accuracy is not uniform.
“Couple and Couple Therapy” for Rescue
His team now relies on another computational chemistry technique, which is also derived from quantum mechanics (called cluster theory) or CCSD (T). “This is the gold standard for quantum chemistry,” Lee commented. The results of CCSD(T) calculations are much more accurate than what you get from DFT calculations, and they can be trustworthy like those obtained from experiments. The problem is that performing these calculations on a computer is very slow, he says, “The amount of scaling is poor: if you double the number of electrons in your system, the calculations will become 100 times more expensive.” So CCSD